Frequently Asked Questions about Real World Evidence / Real World Research
RWR INSIGHTS #1
WHAT DO YOU MEAN BY ‘REAL WORLD RESEARCH’? I’VE HEARD ABOUT REAL-WORLD DATA AND REAL-WORLD EVIDENCE BUT NOT REAL-WORLD RESEARCH.
Great question! Real-world data (RWD) is what you collect. Real-world evidence (RWE) is what you generate from RWD. For us, real-world research is how you collect RWD and generate RWE. It just seems simpler to talk about real-world research studies than to talk about real-world evidence studies. That doesn’t make sense. You don’t conduct an RWE study…you conduct a study to generate RWE. Simple semantics, but it helps to keep us sane..ish.
For those of you who need a referenced definition of ‘Real-World Research’:
REAL WORLD RESEARCH (RWR) refers to the collection of patient-related data in a real-world environment (real-world data), and obtaining clinical evidence (real-world evidence) of the value and potential benefits or risks of the medical products through analysis. The primary research type is observational, but it can also be pragmatic clinical trials (Source – China: Key Considerations in Using Real-World Evidence to Support Drug Development (Draft for Public Review) – Center for Drug Evaluation, NMPA – May, 2019)
RWR INSIGHTS #2
WHAT IS A REAL-WORLD RESEARCH (RWR) STUDY?
Real-world research (RWR) studies fall primarily into two categories: comparative effectiveness (pragmatic1) clinical trials and non-interventional/observational studies2, which can be prospective or retrospective in their design.
References:
1. Pragmatic Clinical Trial (EU definition) = A study comparing health interventions among a randomised, diverse population representing clinical practice, and measuring a broad range of health outcomes. To ensure generalizability, pragmatic trials should represent the intended patients to whom the treatment will be applied as best as possible. For instance, inclusion criteria would be broad (e.g. allowing co-morbidity, co-medication, wider age range, etc.), the follow-up would not be (or not much) interventional and allowing for treatment switching etc. (See also “large simple trials” and “real-world studies”) [Source: imi GetReal Glossary – 25 Oct 2016 Adapted from Schwartz, 1967, Tunis, 2003 & Roland, 1998]
2. Observational Study (USA definition) = A non-interventional clinical study design that is not considered a clinical trial (as per Framework for FDA’s Real-World Evidence Program – Glossary)
RWR INSIGHTS #3
IS GCP APPLICABLE TO NON-INTERVENTIONAL AND OBSERVATIONAL STUDIES?
The short answer is NO.
The longer, more detailed answer, starts with re-phrasing the question…”when designing, conducting, reporting and archiving non-interventional studies (NIS) do we need to comply with ICH GCP?”1
ICH GCP2 = ICH E6, meaning it is guideline number 6 (of 20) in the ICH ‘Efficacy’ series. According to the ICH website: “The work carried out by ICH under the Efficacy heading is concerned with the design, conduct, safety and reporting of CLINICAL TRIALS. It also covers novel types of medicines derived from biotechnological processes and the use of pharmacogenetics/ pharmacogenomics techniques to produce better targeted medicines.”
THE FOCUS OF ICH GCP IS CLINICAL TRIALS, which is why there is reference to “investigational product (IP)”, the “investigator’s brochure (IB)” and research “subjects/trial subjects” who are the recipients of “investigational product(s)”. None of this applies to non-interventional studies, where we assess the effectiveness of routine treatments and clinical practice in patients.
ICH GCP is embedded in clinical trial legislation which is why it is legally enforceable for clinical trials (see EU example below1). The same clinical trial legislation is not applicable to non-interventional studies (usually explicitly stated as such). Meaning? ICH GCP is not applicable to non-interventional studies from a legislative and regulatory compliance perspective.

You are free to follow the principles of ICH GCP (e.g., informed consent process and trial master file content), but you are not legally obliged to do so because it has not been incorporated into, or enforced by, the regional and national regulations applicable to NIS (see below1).

References:
1. GxPs for NIS and Observational Studies – Quick Reference Guide – Is GCP Applicable to Non-Interventional Studies?
Link: https://www.phoenix-rwr.co.uk/gxps-for-nis-and-observational-studies/
2. ICH E6 (also known as ICH GCP) ICH Harmonised Guideline -Integrated Addendum to ICH E6(R1): Guideline For Good Clinical Practice E6(R2), Nov 2016
Link: https://database.ich.org/sites/default/files/E6_R2_Addendum.pdf
RWR INSIGHTS #3.5
IS GPP THE EQUIVALENT OF GCP FOR OBSERVATIONAL STUDIES?
The short answer is NO.
ISPE GPP1 is not embedded in, or enforced through, observational study legislation…anywhere. However, ISPE GPP is referenced in regulator guidance as a recommendation that should be “considered” (see examples below)2.
CANADA – In 2019, Health Canada released guidance on ‘Elements of Real World Data/Evidence Quality throughout the Prescription Drug Product Life Cycle’3. Health Canada references ISPE GPP1 and the ENCePP codes, methodological guides and checklists4,5
EUROPE – The EMA, in GVP Module VIII6 provide direction on the guidelines (GxPs) that ‘should be considered for the development of study protocols, the conduct of studies and the writing of study reports’. These include:
- ENCePP Guide on Methodological Standards in Pharmacoepidemiology4
- ENCePP Checklist for Study Protocols5
- Guideline on Conduct of Pharmacovigilance for Medicines Used by the Paediatric Population7, and
- Guidelines for Good Pharmacoepidemiology Practices of the International Society of Pharmacoepidemiology (ISPE GPP)1
GERMANY – According to the BfArM/PEI joint recommendations for observational studies7, the observation plan should be based on recognized recommendations of scientific or regulatory guidelines. Corresponding specifications can be found, for example in the following guidelines and recommendations:
- “Guidelines for Good Pharmacoepidemiology Practices” (GPP) of the “International Society for Pharmacoepidemiology “(ISPE)1,
- Recommendations of the “ENCePP Guide on Methodological Standards in Pharmacoepidemiology”4, and the
- Guidelines for Good Epidemiological Practice (GEP) of the German Society for Epidemiology (DGEpi)8.
References:
1. ISPE GPP = Guidelines for Good Pharmacoepidemiology Practices of the “International Society for Pharmacoepidemiology
Link: https://www.pharmacoepi.org/resources/policies/guidelines-08027/
2. GxPs for NIS and Observational Studies – Quick Reference Guide – Is GPP the Equivalent of GCP for Observational Studies?
Link: https://www.phoenix-rwr.co.uk/gxps-for-nis-and-observational-studies/
3. Health Canada – Elements of Real World Data/Evidence Quality throughout the Prescription Drug Product Life Cycle, March 2019
4. ENCePP Guide on Methodological Standards in Pharmacoepidemiology
Link: http://encepp.eu/standards_and_guidances/methodologicalGuide.shtml
5. ENCePP Checklist for Study Protocols
Link: http://encepp.eu/standards_and_guidances/checkListProtocols.shtml
6. GVP Module VIII = EMA Guideline on Good Pharmacovigilance Practices (GVP)- Module VIII – Post-Authorisation Safety Studies (Rev 3)
7. Joint recommendations by the BfArM and the PEI on drug observation studies in accordance with Section 67(6) AMG and on the notification of non-interventional safety studies in accordance with Section 63f AMG of December 20, 2019
8. Guidelines for Good Epidemiological Practice (GEP) of the German Society for Epidemiology (DGEpi)
Link: https://www.dgepi.de/assets/Good-Epidemiological-Practice-GEP-EurJ-Epidemiol-2019.pdf
RWR INSIGHTS #4
WHICH GXPs ARE APPLICABLE TO NON-INTERVENTIONAL STUDIES?
GVPs are derived from legislation and legally enforceable for non-interventional studies conducted in the EU (refer to Article 108a of Directive 2001/83/EC as amended)1 – See below…
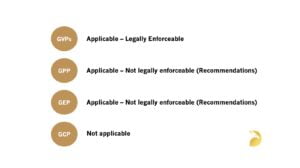
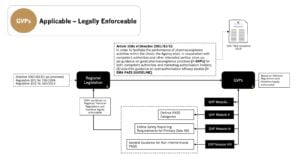
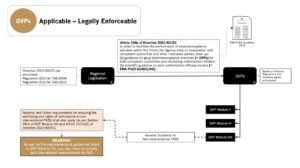
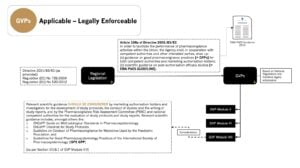
References:
1. GxPs for NIS and Observational Studies – Quick Reference Guide – Which of the GxPs are Applicable to Non-Interventional Studies and Observational Studies?
Link: https://www.phoenix-rwr.co.uk/gxps-for-nis-and-observational-studies/
RWR INSIGHTS #5
WHAT IS THE REGULATORY FRAMEWORK FOR OBSERVATIONAL STUDIES AND NON-INTERVENTIONAL STUDIES?
That’s a very big question! What makes these types of studies complicated is the fact that every country manages and governs them differently. There is no harmonised approach to how these studies are regulated.
So, that was the cop out ‘it depends’ type answer. The figure below provides a high-level overview of the ethical foundations and regulatory framework for non-interventional studies. Just bear in mind this is a top level summary with an EU bias, but it does serve to illustrate the ethical foundation of the Declaration of Helsinki through to the per country regulations.
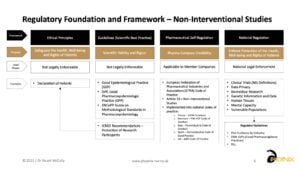
RWR INSIGHTS #6
IS A CLINICAL STUDY THE SAME THING AS A CLINICAL TRIAL?
Not according to FDA and EMA regulatory definitions!1
According to the FDA2, an observational study is a non-interventional clinical study design that is not considered a clinical trial (as per Glossary of Framework for FDA’s Real-World Evidence Program – Dec 2018); and according to the EU Clinical Trials Regulation3, a non-interventional study is a clinical study other than a clinical trial (as per Article 2.2(4) of Regulation EU/536/2014).
Confusion arises when using these terms because according to ICH GCP4, the terms CLINICAL TRIAL and CLINICAL STUDY are synonymous (Section 1.12 of ICH E6(R2)). If you run clinical trials this will probably have little or no impact on you. However, consider those of us who support and manage clinical studies that are not clinical trials e.g., observational studies and non-interventional studies (also known as Real-World Research – RWR or RWR Studies). Our studies are NOT the same or even similar to clinical trials, especially when it comes to the regulatory requirements we need to comply with. For a start, we aren’t governed by regulations that have their basis in ICH GCP. So…why add terminology to clinical trials guidance document that impacts observational studies and causes confusion, misdirection and horror(!) to those of us who already have our hands full trying to address the regulatory complexity of ‘not clinical trials’.
I can illustrate the point more clearly if I ask the question differently – Are observational studies the same as clinical trials?
I posed this question to my LinkedIn network early in 2021 and received the following clear response = No, observational studies are not the same as clinical trials.
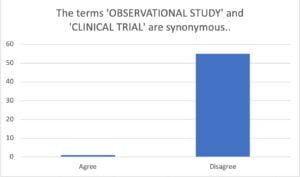
So, same audience…rephrased question/statement…
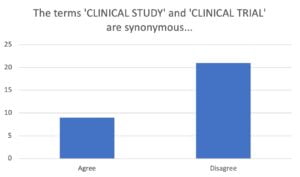
The terms ‘clinical study’ and ‘clinical trial’ are not the same, but there is obviously confusion as to whether they are different, largely thanks to the misrepresentation of the terms by the ICH team in ICH GCP. The impact of grouping them together is that it causes confusion at the study design, approvals/submissions, conduct, and reporting levels.
Take Home Point = The terms ‘Clinical Trial‘ and ‘Clinical Study‘ are not synonymous.
References:
1. Clinical Trial vs Clinical Study – Synonyms or Significant Differences
Link: https://www.phoenix-rwr.co.uk/clinical-trial-vs-clinical-study/
2. Framework for FDA’s Real-World Evidence Program – Dec 2018
Link: https://www.fda.gov/media/120060/download
3. EU Clinical Trials Regulation (Regulation EU/536/2014)
Link: https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/reg_2014_536/reg_2014_536_en.pdf
4. ICH E6 (also known as ICH GCP) ICH Harmonised Guideline -Integrated Addendum to ICH E6(R1): Guideline for Good Clinical Practice E6(R2), Nov 2016
Link: https://database.ich.org/sites/default/files/E6_R2_Addendum.pdf
RWR INSIGHTS #7
WHAT ARE THE ARCHIVING REQUIREMENTS FOR NON-INTERVENTIONAL STUDIES?
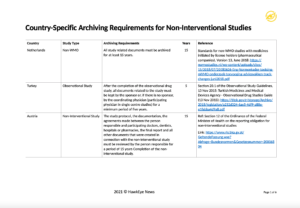
Most countries do not stipulate any legal requirements for document retention and archiving of non-interventional study documents. The following table provides a quick overview of countries that do have explicitly stated archiving requirements for NIS/ observational studies.
See example. Download the full FREE PDF copy of “2021 NIS Country-Specific Archiving Requirements for Non-Interventional Studies” below:
RWR INSIGHTS #8
IS THERE A TRIAL MASTER FILE INDEX DESIGNED FOR OBSERVATIONAL STUDIES AND NON-INTERVENTIONAL STUDIES?
There is indeed! It was published in 2020 by the TMF Reference Model team1.
There are two forms of the ‘real-world study document index (RWS-DI)’ available:
- Distilled RWS-DI2
- This is a Document Index (read ‘TMF’) specifically intended for use with Real World Studies that has been adopted from the TMF Reference Model.
- This is the distilled version of the RWS-DI that is posted on the TMF Reference Model website (see link below).
- We call this the “distilled” version because it doesn’t contain the guidance text, glossary or information on the scope of the Document Index.
- Detailed RWS-DI3
- Think of this as the raw data (Distilled RWS-DI) plus all the additional guidance and instructions for use. Useful for anyone who wants to understand the scope of the Document Index and Real-World Study context of the Sections, Zones and Artifacts.
- The ‘Detailed’ RWS-DI includes:
- The Distilled RWS-DI
- + Guidance and Context on How to Use the Document Index
- + Details of the Types of Real-World Studies that the RWS-DI was designed to support
- + A Glossary of Terms
References:
1. TMF Reference Model – Real World Study Document Index – Provides a proposed Document Index for use on real-world studies, based on the TMF Reference Model for clinical trials (v1.0 Approved 29-July-2020)
2. Distilled RWS-DI
3. Detailed RWS-DI
RWR INSIGHTS #9
IS AN OBSERVATIONAL STUDY THE SAME AS A NON-INTERVENTIONAL STUDY?
Yes, no and it depends…
It is a confusing area! Compared to the ‘maturity’ of clinical trial legislation, which fundamentally traces its global harmonisation from the 1996 publication of ICH GCP…Real-World-Research (RWR) is still in its ‘legislative’ infancy…which is why there is currently no single recognised ‘book of terms and definitions’ or glossary. Each country creates their own terms or borrows and adapts from other countries1.
The result? A complex confusion of country-specific terms and definitions…that all mean the same thing…Almost!

From a regulatory compliance perspective, it’s important to remove the ‘white noise’ of the methodological and study design terminology and focus on the regulatory terms, definitions and classification for each of the countries you plan to conduct your study in.
The Phoenix-RWR websites has a couple of useful tools that can help you beat the confusion of terms and definitions:
- Table of Regulatory Definitions and Classifications of Real-World Research Studies
- Glossary of Real-World Research Regulatory Terms and Definitions
References:
1. Real-World Research Studies – A Complex Confusion of Country-Specific Terms and Definitions…That All Mean the Same Thing…Almost
Link: https://www.phoenix-rwr.co.uk/real-world-research-studies/
2. Phoenix-RWR – Table of Regulatory Definitions and Classifications of Real-World Research Studies
Link: https://www.phoenix-rwr.co.uk/real-world-research-studies/
3. Phoenix-RWR – Glossary of Real-World Research Regulatory Terms and Definitions
Link: https://www.phoenix-rwr.co.uk/useful-resources/phoenix-glossary/
RWR INSIGHTS #10 HOW DO I FIND OUT WHAT THE NATIONAL REQUIREMENTS ARE FOR MY OBSERVATIONAL STUDY?
I’ve always talked about the ‘complex simplicity’ of non-interventional studies. NIS are, by design, simple. The challenge comes from the local regulatory differences that impact the classification categories, study start-up requirements and operational aspects of NIS. For studies that are so simple (by design) and so low risk (the patients are already taking the drugs), they can be challenging to run. But they don’t need to be…
STEP #1 = Determine the High-Level Regulatory Classification for your Proposed Research (What is it?)1
[1] Safe Baseline – All Countries Define and Regulate Clinical Trials
[2] Comfortable Starting Point – Start by Determining Whether your Study is a Clinical Trial
[3] Study Classification and Regulation – If it isn’t a Clinical Trial, then What is it and How is it Regulated?
The regulatory classification is important because it decides what regulatory pathways you need to comply with e.g., clinical trial (global), imposed post-authorisation safety study (Europe), non-interventional study of a new drug (India), post-marketing requirement (USA), AWB (Germany).
The following decision tree provides a generic overview of the classification considerations for these types of studies.
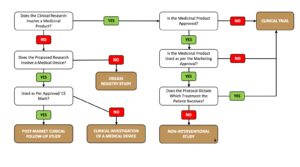
STEP #2 = Determine the Local Regulatory Classification for your Proposed Research (What is it Locally?)
If we take India as an example, the approval requirements differ for non-interventional studies conducted with ‘New Drugs’ versus those conducted with ‘Old Drugs’. Classification is based on risk and is explained in Rule 2(w) of the New Drugs and Clinical Trials Rules, 2019. The English translation begins on page 147. The Fifth schedule, which deals with the regulatory requirements for these studies, begins on page 228.
Refer to the classification decision tree below…
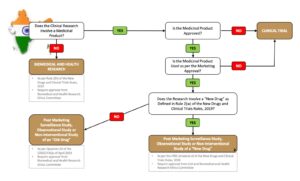
STEP #3 = Determine the Local Regulatory Start-Up Requirements for your Study (What Do I Need to do?)
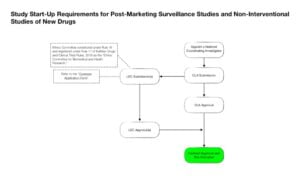
References:
1. NIS Considerations – Conquering the Complex Simplicity of Non-Interventional Studies
Link: https://www.phoenix-rwr.co.uk/nis-considerations/
RWR INSIGHTS #11
WHAT ARE THE REQUIREMENTS FOR NOTES TO FILE (NTF) FOR OBSERVATIONAL STUDIES?
The Real-World Evidence Study Master File Team1 has received feedback that Notes to File (file notes) are not listed in the artefacts on the RWE SMF Index2.
When creating the RWE SMF, the team deliberately focussed on artefacts required by regulation or otherwise considered relevant to RWE studies. There is no regulation that mandates or requires NTF and so the RWE SMF Team decided not to include NTF as an artefact for routine inclusion.
Where so desired, NTF can be added as an artefact in the same way as it is possible to add artefacts to the standard TMF Reference Model. NTF can be useful in exceptional circumstances where required to
- explain exceptional situations;
- define the root cause;
- evaluate the impact;
- describe actions taken to correct the situation; and
- document remedial action(s) taken to prevent recurrence.
However, rather than provide clarity, recording “after-the-fact” matters in NTF potentially
- magnifies sometimes insignificant issues;
- adds confusing, superfluous, or extraneous information;
- highlights ineffective practices, poor performance, and unresolved problems; and
- reveals detrimental evidence regarding inadequate study conduct and monitoring.
And, of course, NTF should never be used to justify
- failure to perform due diligence;
- failure to understand or comply with regulatory requirements or the protocol; or
- missing documents (if an artefact cannot be collected, the NTF does not replace it).
To prevent a culture overly sensitive to- even fearful of- risk, organisations should establish “golden rules” i.e. a framework of SMF policies, standard operating procedures, work instructions, guidelines, or conventions on NTF to
- anticipate (and be prepared for) questions from auditors and regulatory inspectors;
- ensure that NTF are created only in relation to significant issues that have a material impact on the conduct of the trial, the quality of the data produced, or patient safety; and
- avoid being drawn into time-consuming attempts to retrospectively apply “corrections” that might potentially prove counterproductive.
It is important to adopt a pragmatic approach to NTF creation that balances effort required against risks presented: and to guard against hoisting red flags by providing auditors and regulatory inspectors with the rope with which to hang yourself.
About the Author: Russel Joyce
Russell is Director and Principal Consultant of Heath Barrowcliff Consulting Ltd3, Director of the Health Sciences Records and Archives Association (HSRAA), Steering Committee Member of the TMF Reference Model, and Project Lead for the Real World Evidence Study Master File Index.
References:
1. Real-World Evidence Study Master File Team
Link: https://studymasterfile.com/our-team/
2. RWE SMF Index
Link: https://studymasterfile.com/rws-di-2/
3. Heath Barrowcliff Consulting Ltd